|
پروفسور محمد حسین سلطان زاده
استاد
دانشگاه علوم پزشکی شهید بهشتی
متخصص کودکان ونوزادان
طی دوره بالینی عفونی از میوکلینیک آمریکا
دبیر برگزاری کنفرانس های ماهیانه گروه اطفال
دانشگاه علوم پزشکی شهید بهشتی
|
اقای دکتر
شاداب صالح پور
فوق تخصص غدد ومتابولیک اطفال
به
اتفاق اعضای هیئت علمی
بیمارستان لقمان حکیم
معرفی کیس توسط
|

اقای دکتر دکتر زندی
رزیدنت بخش اطفال بیمارستان لقمان

معرفی بیمار:
A 6 year old child with seizure disorder and progressive
neuro-developmental regression
The boy had been born to a 22-year-old woman by spontaneous vaginal delivery
after an uncomplicated, full-term, second pregnancy. He sat up at nine months of
age and walked at one year. At 18 to 24 months, his vocabulary was noted to be
limited, his speech difficult to understand, and his intellect inferior to that
of his 7-year-old brother at those ages. His only illnesses were repeated bouts
of tonsillitis, which led to a tonsillectomy at the age of four years. Between
the ages of two and five years, he fell with increasing frequency. At four years
of age, he was able to use the toilet on his own and to play soccer. During the
two years before admission, his cognitive and motor functions declined.
Eight months before admission, he had an episode in which his arms stiffened and
extended and he fell backward; he recovered after a few seconds. Such episodes
began to occur three or four times daily during waking hours. Occasionally, he
was incontinent of urine or stool during the episodes or had a general loss of
muscle tone. During subsequent months, he chewed and swallowed increasingly
slowly, required soft food, and lost 5 kg in weight. He interacted progressively
less with his mother. Eventually, the attacks occurred 5 to 10 times daily; he
became fearful of falling backward, and instead of walking, he crawled.
A magnetic resonance imaging (MRI) examination of the brain, performed elsewhere
on an uncertain date, revealed diffuse cerebral and cerebellar atrophy and a
poorly defined band of increased signal intensity in the periventricular white
matter on T1-weighted images; the lateral ventricle and the cisterna
magna were slightly enlarged, but the cerebellum appeared intact.
The boy's IQ was 50. Six months before admission, treatment with valproate
sodium–valproic acid reduced the frequency of the episodes, but the patient's
family was concerned about the possible toxic effects of the medication on his
liver. Carbamazepine was substituted, but his condition did not improve, and he
became somnolent. Treatment with valproate sodium–valproic acid was resumed, but
his parents reduced the dose from 800 mg to 200 mg daily. He was referred to
this hospital.
The family resided in a Middle Eastern country. His parents were first cousins.
There was no family history of neurologic disease.
General physical examination revealed no abnormalities. On neurologic
examination, the boy was alert; he reached for toys more often with his right
hand than with his left. He was able to follow simple, one-step commands. His
speech was barely intelligible. Ophthalmologic examination revealed moderate
limitation of the elevation of the eyelids; probable concentric reduction of the
visual fields, with reduced visual acuity; retinal pallor; and bilateral,
cherry-red spots. The optic nerves appeared normal. He had slow saccades and
difficulty initiating saccades. Other cranial-nerve functions appeared to be
intact. Muscle strength was normal. Axial muscle tone was normal; appendicular
tone was slightly increased. Sensation of a pinprick was intact. The deep-tendon
reflexes were +++, with bilateral Babinski signs and clonus at both ankles.
Tremors were evident when the boy sat upright, stood, or reached for an object.
He was unable to stand for more than 3 or 4 seconds or to sit for more than 30
seconds before falling backward. With assistance, he walked with a wide-based,
moderately ataxic, spastic gait.
The urine was normal. The pyruvate level in the blood was 0.82 mg per deciliter
(93 μmol per liter). A test for resistance to activated protein C was negative.
A cranial MRI examination
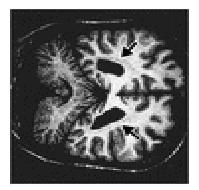
Coronal T1-Weighted MRI Scan at the Level of the Posterior Aspect of
the Lateral Ventricles.), performed without the administration of contrast
material, disclosed a poorly demarcated, periventricular, band-like signal that
was present bilaterally and that was isointense in relation to cortical gray
matter on all imaging sequences. There was slight prominence of the ventricles,
cisterns, and sulci. Intracranial flow voids and the orbits appeared normal.
A lumbar puncture was performed which was normal for glucose, protein, IG, and
lactate levels. An electroencephalographic study showed slight background
slowing of theta waves, at 50 to 70 μV; frequent bursts of a frontally
predominant, 2-Hz spike and slow-wave discharges at 500 μV; and slowing, with
frequent single spikes and slow-wave discharges, in occipital areas,
particularly on the left side. The frequency of the spike–wave activity
increased with sleep. Median-nerve somatosensory evoked potentials had a central
conduction time of 9 msec, with an N19–P22 amplitude of 20 and 30 μV (on
left-sided and right-sided stimulation, respectively). On strobe-flash testing,
visual evoked potentials had a positive component (22 to 24 μV at about 90 msec).
Peripheral-nerve conduction velocities in the legs were normal (40 cm per
second).
تشخیص شما چیست ؟
What is Your Diagnosis ?

